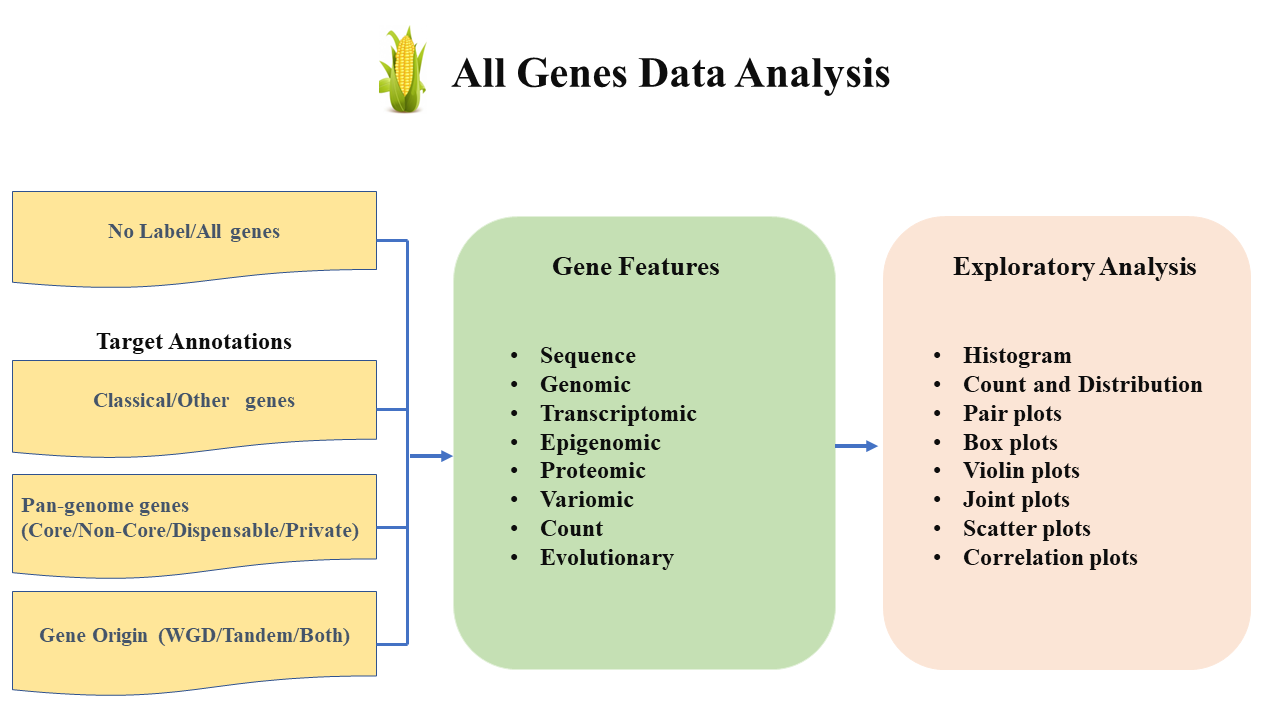
What is All genes data analysis?
- The Maize Feature Store (MFS) "All Genes Analysis" is designed to assist users in visualizing the distribution of diverse gene-based features for either All genes without label (No Label) or exploring the relationships between the gene-based features and gene annotations: (Classical/Other, Pan-genome, Gene Origin) categories offered within the MFS platform.
- For more details on the usage or understanding of the "All Gene Analysis" module, go through the flow diagram or the video tutorial attached below.

Lorem ipsum
Omics datasets come in a diverse range, scale, and follow their own statistical distributions as they are collected from disparate sources, therefore data standardization becomes crucial for omics datasets. Outputs generated from non-standardized features are often skewed, deviated, and filled with outliers and anomalies. Thus, the advanced exploratory analysis such as Dendrograms, Hierarchical Heatmaps, Hierarchical Scatter plots, Heatmaps and PCA analysis demands high-level data preprocessing and normalization to balance out disproportionate weights across multiple variables. Data normalization transforms the multiscaled data all to the same scale, thereby improving the stability and performance of the learning algorithm. The Maize Feature Store application allows for the normalization of omics numerical features by centering the features with their mean and the standard deviation between 0 and 1 using the most common normalization method called Z-score normalization. In standardized z-score normalization, each feature is normalized as Z = ( X - X' ) / S, where X, X' and S are the feature, the mean and the standard deviation respectively.
- Data Tables
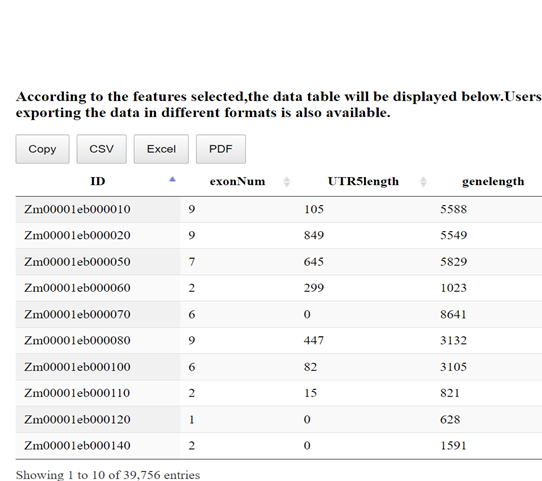
- Data Visualization
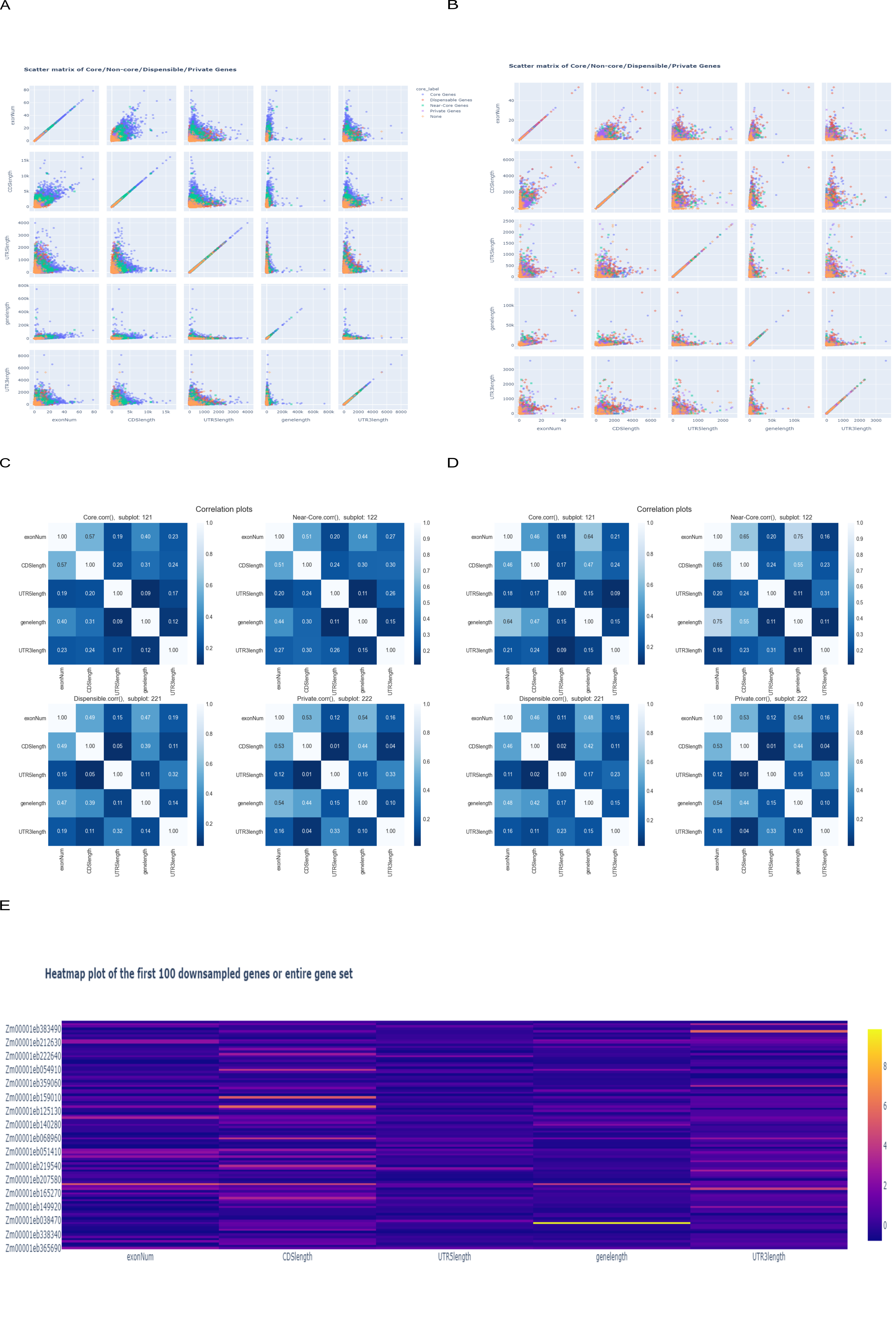
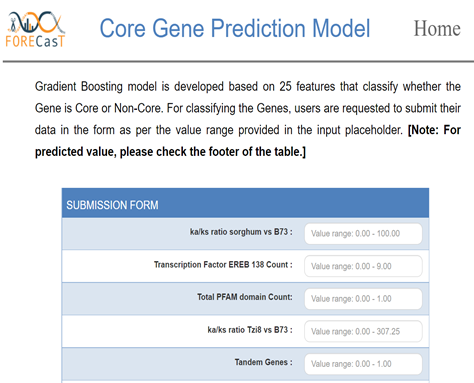
- Data Modeling
- What is common among these genes in a given class?
- What are the relationships between: gene phenotype and gene length, copy number, expression levels and patterns, epigenetic markers, cross-species conservation, and SNP densities ?
- Apply machine learning methods to predict important biological classifications.
- Provide tools to discover if genes within a class have distinct features and utilize it for constructing within- and cross-species prediction models.